Sequence search
Landing page
The sequence search (accessed by following the ‘Sequence search’ link from menu bar) provides a search against a catalogue of predicted peptides.
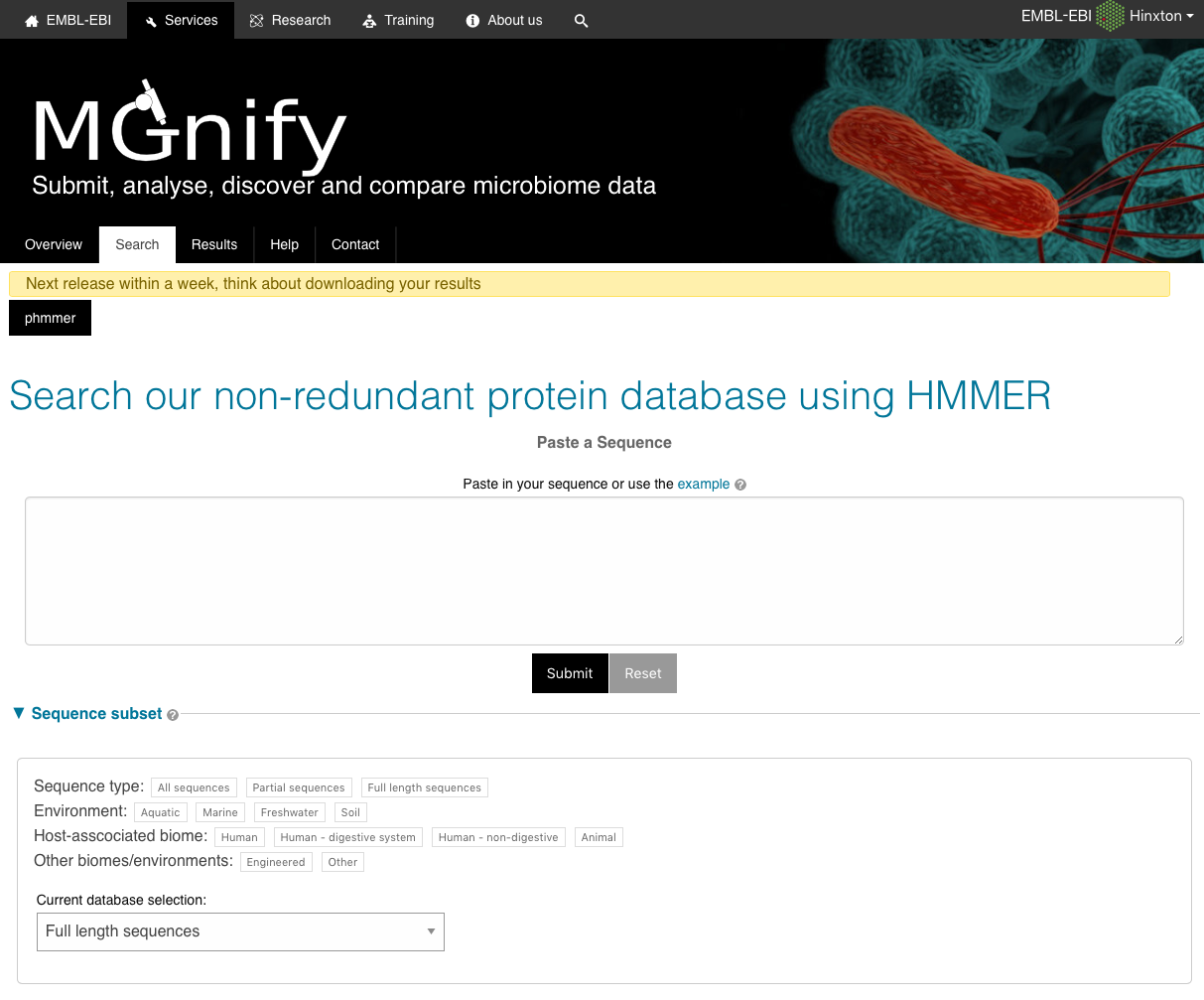
These sequences comprise a non-redundant set of proteins predicted from contigs that have been assembled from sequencing runs. The HMMER search engine has been adapted to provide fast searches against this database. The results can be linked back to the sample and run from which the peptide was derived and also to sequences with an exact match in the UniProtKB database.
Owing to the large size of the database we are not able to offer a search against the full set of proteins. Instead, we have applied a clustering algorithm which groups sequences into clusters based on similarity. The clusters each have a ‘representative sequence’ and it is these that are offered in the search, though the full set of proteins may be obtained from our FTP server.
The search takes a FASTA-formatted amino acid sequence.
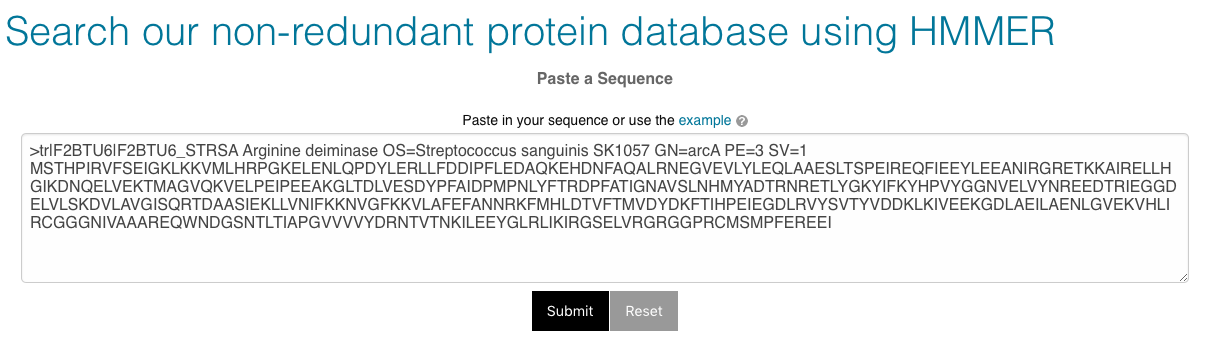
You can search against all of the sequences in the database (‘All’), or restrict your search to full length sequences, or partial sequences only (see Partial and full length peptides). Alternatively, you may choose to search from a subset of environments or biomes. Sequences observed in multiple runs may be found in more than one biome. ‘Other’ sequences are those found in none of the other environment or biome categories.
Result page
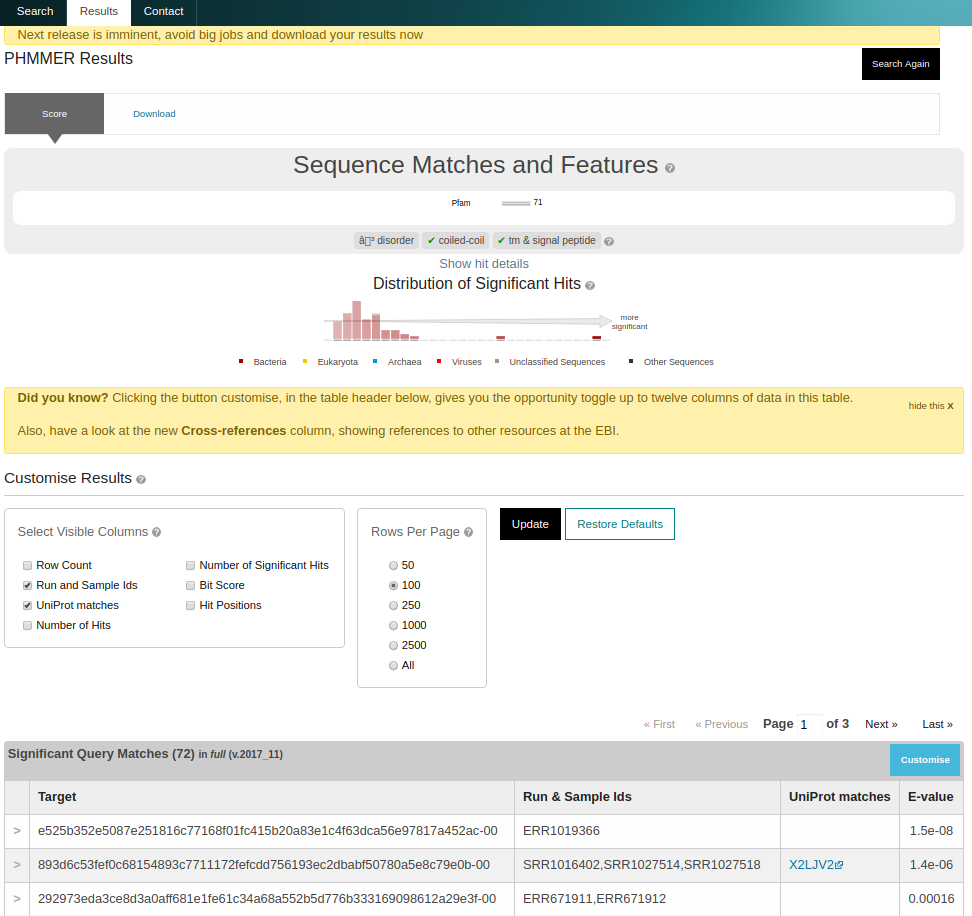
On completion, a list of matching sequences is shown in order of E-value significance. All sequences in the database have a stable MGYP accession. Additional columns, such as the mapping to UniProtKB identifiers, can be enabled by clicking ‘Customise’ on the results page and checking the appropriate boxes.
Build process
The database is updated periodically and is created as follows:
Short reads from runs are assembled into contigs using an assembler, such as metaSPAdes
Contigs are filtered by length (minimum 500 base pairs)
Peptides are predicted using the Prodigal gene caller
Resulting peptides are made non-redundant to produce a set of unique sequences
Sequences are mapped back to MGnify run and sample accessions and annotated with biome(s)
Matching sequences in UniProtKB are identified
Sequences are clustered using MMseqs2/Linclust
Each update (versioned using the release year/month) is cumulative and uses all predicted peptides available at that time.
Partial and full length peptides
In common with some other protein coding sequence predictors, Prodigal provides an indication as to whether a gene is full length or extends beyond the contig. This is recorded as two digits (one for each end of the sequence), each of which is either 0 (the gene is encoded within the contig) or 1 (it extends beyond). Thus a full length sequence is described as ‘00’ and a partial as ‘11’. The values ‘10’ or ‘01’ are used for the cases where the gene is truncated only at one end.
>seq_1 # 3 # 371 # 1 # ID=1_1;partial=10;start_type=Edge;rbs_motif=None;rbs_spacer=None;gc_cont=0.501
SEGCEYLAAYLDKRIASGETINESSAVMTLSQGYLMKGRNKDAGKKFITTPAITKEIREA
QT
>seq_2 # 4738 # 5193 # -1 # ID=1_9;partial=00;start_type=ATG;rbs_motif=None;rbs_spacer=None;gc_cont=0.568
MSAYWYAVIWGGSFGAVLAAAGPRFRKAIPAIRGRMKNSIKWSTSAKAINGISWAGPFAA
QT
>seq_3 # 7546 # 8232 # -1 # ID=1_11;partial=00;start_type=TTG;rbs_motif=GGAG/GAGG;rbs_spacer=5-10bp;gc_cont=0.541
MKKKVLSIQNIACETLGTLEGMFRKDGLEVENVSAQEGGIPIKSSEYSAVVVLGGPMAVY
QT
>seq_4 # 32 # 103 # -1 # ID=37115_1;partial=01;start_type=Edge;rbs_motif=None;rbs_spacer=None;gc_cont=0.542
WILDGIDIDAMIRHPVRQYQIAGAvailability
As well as searches via a web server, we provide all data for download from our FTP server. This includes the sequence database (separate fasta files for the full database and cluster representatives); run, sample, biome, Swiss-Prot and TrEMBL mappings; the partial status of the sequences and counts of the number of times each sequence was observed in the database as a whole.
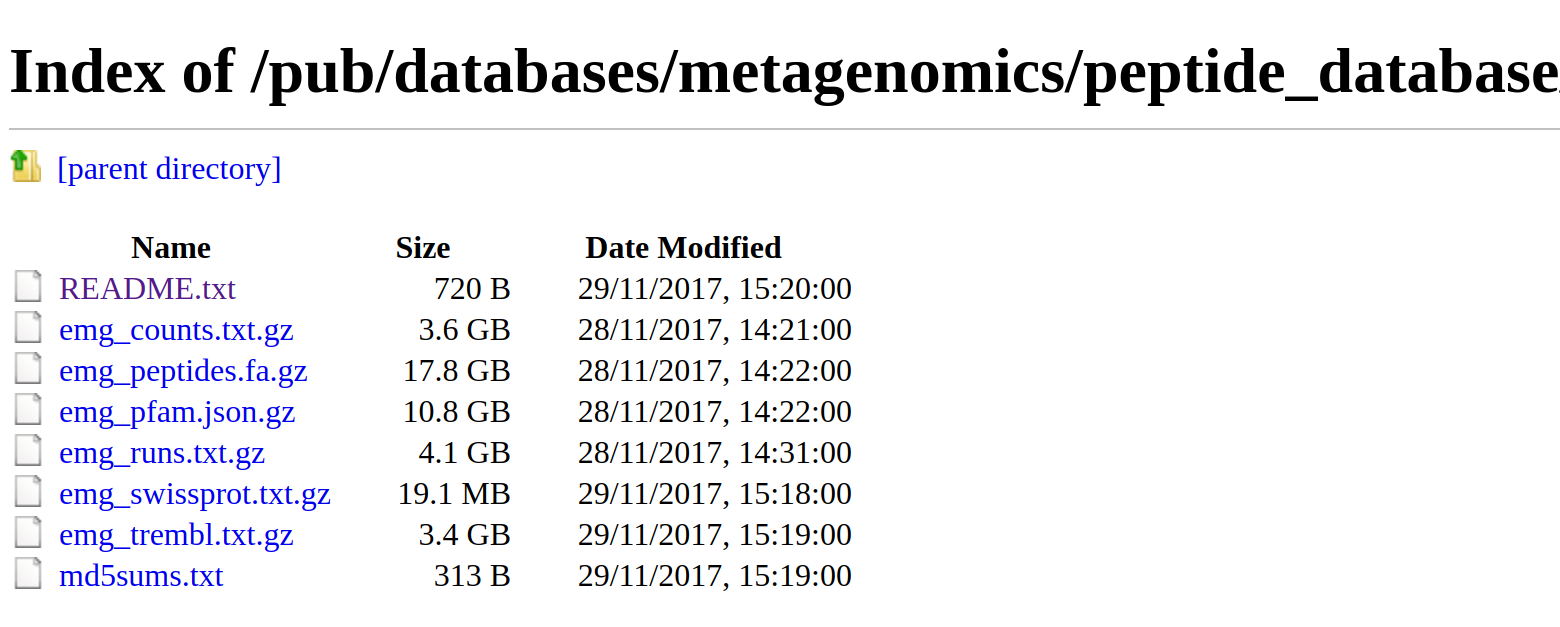
Further information
Full documentation regarding the HMMER webserver is available. Note that some of the documented features (such as the taxonomy view) are not relevant to the peptide search and are therefore disabled. If there are additional features or feedback on this search service, please get in contact with us.
Citation
@online{2024,
author = {, MGnify},
title = {Sequence Search},
pages = {undefined},
date = {2024-03-12},
url = {https://docs.mgnify.org/src/docs/sequence-search.html},
langid = {en}
}